Screening Assistant 2 v1.0
Import workflow
Last edited: 18/11/2011
By: VLG
Sourceforge: SA2 website
Help: SA2 forums
Documentation index
This page describes the detailed procedure used to import a new molecule (and eventually some properties) in the database.
Table of Contents
- Database representation - how your molecules are represented in the database
- Duplicates - how SA2 deals with duplicates molecules
- Pre-processing - are your input molecules changed by SA2 ?
- Workflow - the detailed import procedure
Database representation - top
The first important thing to know is that screening assistant represents the structure of your molecule using the MDL MOL-formatted connection table. All further information in each SD entry (basically everything that comes after the M_END flag) will be skipped.
Even more importantly, absolutely no modification will be performed on your original connection table! The connection table will be stored as provided, and SA2 don't even add hydrogens, remove salts or perform any other action on your original entry (at least in the current default version of SA2 - see next section). The standardization of each molecules is therefore up to you!
Why is that so? Well, the standardisation of molecules is usually laboratory-specific, and even sometimes problem-specific (e.g. for descriptors calculation). Thus, we decided that SA2 will basically percieve molecules just as provided by you, with absolutely no further processing.
It is however still possible to include your own standardization procedure directely within SA2, but this is to be documented (see the Pre-processing section), and you will need a JAVA-programmer to do so.
Duplicates - top
SA2 stores unique molecules. We use the IUPAC InChI Identifier to represent each molecule and detect a duplicate molecule. Thus, tautomers will be detected as duplicated molecule (at least as far as the InChI algorithm can detect them...). However, a charged carboxilic acid will be detected as a different molecule compared to its neutral form.
SA2 also takes into account stereochemistry when computing the InChI.
Pre-processing - top
We told you previously that SA2 do not change your molecules in any partucular way. This is clearly true in the current default version of SA2. However, SA2 makes it possible to define (if you know java programming ;)) so-called transformers. A transformer will generated a new SD file based on the original one, and thus eventually perform some custom, developper-defined standardization on each molecule.
As already said, there is currently no transformer available in the default version of SA2, but it's worse to note their existence if you know JAVA, and as they might come up in future major release of SA2 as optional features.
Workflow - top
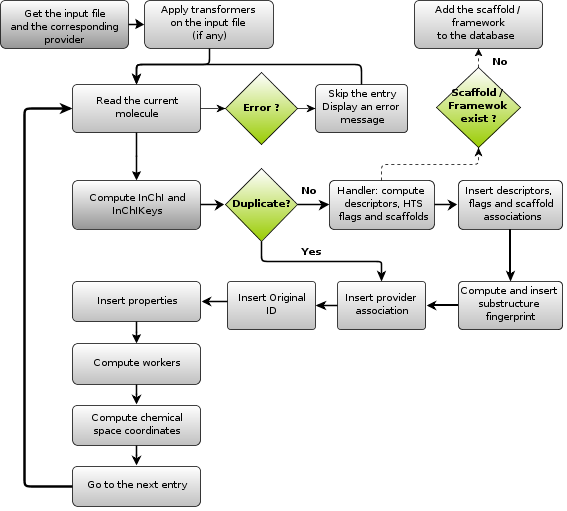
Lets now enter in more details on the import process. Before processing each molecule successively, we do the following:
- Count the molecules in the input file
(can be time-consuming depending on the size of your input file)
- Apply transformers
(not used in the default version)
- Compute InChI and InChIKeys on the entire input file
- Check integrity of results
If the number of output inchi is different than the number of input molecules, throw an error.
Once done, we loop over each SDF entry ($$$$ separated), and for each entry, we do the following:
- Check duplicates
Determine if the molecule already exists (using the InChIKey and InChI identifiers)
- If the molecule does not exists yet
- Compute and insert the basic descriptors.
If the molecule has more that 100 atoms, skip it.
Note that you can change this number.
- Compute and insert each HTS flags (if asked)
- Compute the scaffold and the framework of the molecule. Insert both if they do not exist yet (using InChI as well).
- Compute the substructure fingerprint.
- Compute and insert the basic descriptors.
If the molecule has more that 100 atoms, skip it.
- Associate the molecule and the Provider
If this molecule was already available for this provider, increment the number of duplicates for this association.
- If asked, associate the molecule with the selected library
- Insert the original ID of the molecule
If the ID already exists, do nothing.
- If any, insert the properties in existing tables
If these properties were already available for this molecule, they will be updated.
- If any, insert the properties in new tables
The new tables will be created on the flight. If these properties were already available for this molecule (duplicates in the same input file), they will be updated.
At this point, if any error has occured, the molecule will not be imported and an error message will be displayed in the output window.
- If the molecule is not a duplicate, perform additional operations (selected
workers). If any worker fails, the molecule
will still be imported but the data associated with the worker will not be available.
-
Compute DRCS (2D PCA chemical space) coordiates.
If you do not compute or import the descriptors associated with each available chemical space (i.e. CDK descriptors and MOE descriptors), the coordinates will not be computed and you will not be able to use the already existing chemical spaces.
Here is a graphical outline of the import workflow: