Screening Assistant 2
Searching molecules
Last edited: 04/11/2011
By: VL
Sourceforge: SA2 website
Help: SA2 forums
Documentation index
In this page, you will learn how to search for specific molecules in your database. There are various ways of searching molecules in SA2, including similarity, substructure, exact structure... We will describe how to use them, a point-out their advantages and limitations.
Table of Contents
Name search - top
You can search molecules using a name pattern that will be used to match the original IDs (the ID field that you had selected when importing your molecules). To do so, a text field is available on the top toolbar:

To search for molecules using this text field, simply enter the name, or part of the name of your molecule(s), and either press enter, or the button located on the left of the text field. Once done, all molecules that match the entered name will be retrieved and displayed in a results frame.
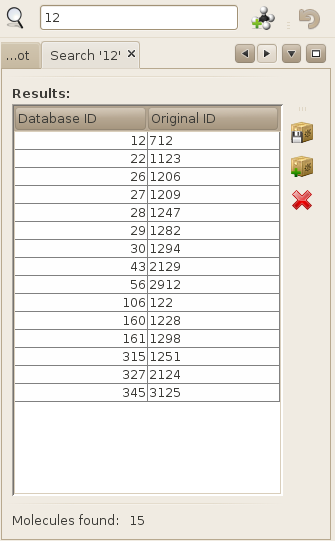
You can then perform all operations available on the right toolbar, such as saving your results in a new library, adding your results to an existing library, or simply deleting your molecules from the database.
Note that regular expressions are not supported. However, substrings are. If your type 'a' as a search query, all molecules that contain the 'a' letter in their name will be retrieved.
Exact structure search - top
You can also search for a specific structure by sketching it and searching the
sketched structure in the database. To do so, either use the toolbar shortcut
 , or the menu
Compute->Exact Structure search. You will be asked to draw your
query molecule in the available sketcher (JChemPaint by default).
, or the menu
Compute->Exact Structure search. You will be asked to draw your
query molecule in the available sketcher (JChemPaint by default).

To perform the exact search, SA2 will compute the InChI identifier of your query, and search for it in the database. The search is therefore quite strict: although tautomers are handled, if you e.g. search for a carboxylic acid in its neutral form and that you enter the negative form, you will not retrieve your molecule.
The results will be displayed in the same window as previously described, or a message will be displayed if the molecule could not be found or if your query molecule could not be processed by the InChI program.
Similarity search - top
Looking for similar molecules is particularily usefull in many aspects of drug discovery. The interesting part of similarity search is that there is no absolute definition of the similarity between two molecules, neither there is an absolute way of quantifying this similarity. Thus, you will need to choose the description of your molecules, and the metric quantifying the similarity between two descriptions, to perform similarity search. This choice will usually be guided by your own specific requirements.
Let's described a bit more both aspects and put them in the context of using SA2.
Description
In a standard similarity search, molecules are encoded using Fingerprints, which encodes various structural, topological or pharmacophoric features of each molecule in a usually large bitstring. Each feature is thus encoded as 0 (if present) or 1 (if absent) in a specific position of the bitstring, and sometimes even by a number representing the feature count instead of its presence / absence. In SA2, we only use binary fingerprints.
In the current version of SA2, similarity search can be performed using any available fingerprint. In addition to the fingerprints that can be calculated through workers, you also have the possibility to create a new fingerprint (under the hood, a new storage capability in the database), and subsequently import you fingerprints as illustrated in the quickstart guide.
Of course, when a fingerprint (newly created) cannot be computed directely within SA2, you will not be able to perform similarity searches using a query molecule that does not exists yet in the database. Read more about that in the Properties section of the documentation.
Metric
Once you get a fingerprint for each of your molecules, you have to quantify the similarity between your query molecule (fingerprint!) and all molecules available in the database. To do so, you need a metric. In SA2, the Tanimoto metric is currently available by default, but there are many more! Some other metrics (cosine, Tversky) will be added later, as the API has been designed to add very easily new similarity metrics.
Example
We will now search for compounds similar to an existing molecule. You need a valid and populated SA2 database to follow this example. If not already, follow the quickstart guide.
- Open any of the tables available and left click on a molecule of your choice. Alternatively, you can also click on the 2D structure view after selecting a molecule in any other view, and select the Similarity search (FP) menu.
-
Or ou can draw your own molecule in a sketcher and compute the search:
Compute->Similarity search, but as pointed previously, you will be able to use only fingerprints that can be computed within SA2.
In both cases, the following window will be opened (with eventually your query molecule drawn in the sketcher).
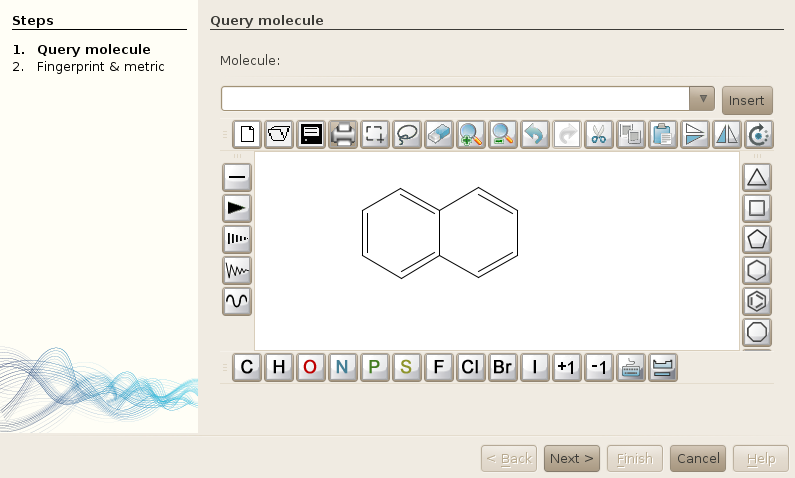
The next step will be to select your fingerprint, the similarity metric, and the bounds of this similarity metric. Remember that if you select a fingerprint that has not been calculated (using workers), or imported, you will not find anything :).
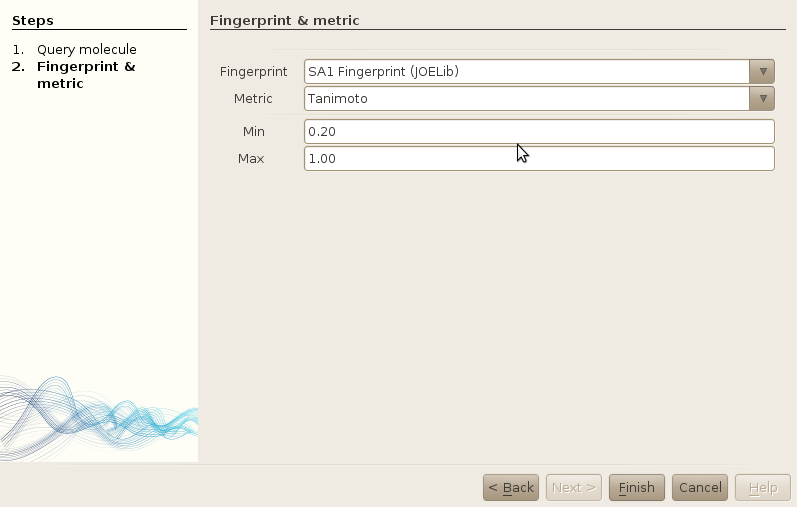
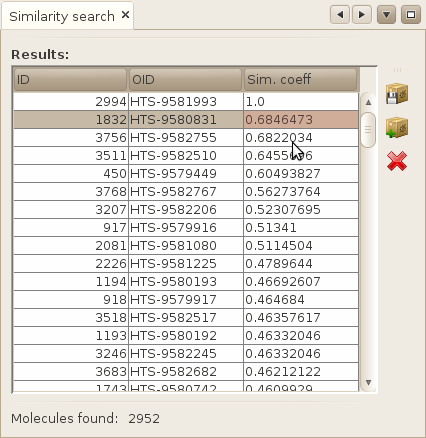
Click on finish to launch the search. The results will appear in a specific search results window, ordering your molecules by decreasing similarity value.
Substructure search - top
Substructure search is a basic feature that a chemoinformatics software should have. Yet, it is not that easy to implement a fast, efficient and flexible substructure search. Let's explain how it is performed within SA2.
First of all, you will have to enter your query substructure, either based on an existing molecule (left clic on a structure in any of the tables), or from scratch. Use the Compute->Substructure search menu to open the following window:

Next, you will have to select the substructure engine to use to perform the matching. Why do we need a substructure engine ? Well because although SA2 uses a pre-filtering step to remove molecules that cannot match the required substructure, the remaining molecules still have to be tested somehow, and this is the job of the substructure engine.
Currently, there are two engines available. They perform both almost equally well in terms of performances, with a little advantage to the Indigo engine (which is selected by default). Keeping the default engine is still a good idea ; using another engine can be usefull mostly for developers willing to compare different matching algorithms, and see how they handle exotic queries.
Optionally, you can restrict the search to a particular library. Once configured, you can run the search, which will proceed the following steps:
- Get the query substructure and the substructure engine
- If the substructure correspond to an existing scaffold, retrieve all molecules associated with it. Otherwise, do nothing... :)
- Filter-out the remaining molecules that cannot match this substructure (using a specific fingerprint and query)
- Loop over each remaining molecules
- Add new hits as they come to the results window
Note that although the search has been dramatically improved since the 1.0.* versions, it is still limited in some ways. For example, if you search all molecules containing a benzene, well, there are potentially millions of match (for large databases :)), and it will take some time to retrieve all of them... The scaffold alows to retrieve very quickly a part of the results, but still. The more specific is your query, the more rapid will be the search.
The results are displayed as soon as they are retrieved, so you won't have to wait for the entire process to finish before looking at the first results.
SMARTS search - top
SMARTS search can be performed in the database or in a particular library. In both cases, you must be aware that the search is currently performed by scanning the entire database (or library), and is therefore quite slow for large databases (you'll need around 45 seconds to search within a database of 15 000 molecules with the fastest SMARTS engine).
By default, the SMARTS search window is opened and reduced on the left sliding side of the main window. You can open it using the menu bar: Compute->SMARTS search

Using this window, you simply have to enter you SMARTS query in the dedicated text field. Optionally, and thanks to a wonderfull web service created by K. Schomburg and described in [1], you can get a visual depiction of your query by clicking on the View button. Very usefull, but you will need an access to internet as the service is only available online.
You can also check that your SMART query is supported by the selected engine using the Check button.
Finally, you can eventually select a different SMARTS engine. Note that the default engine (using the Indigo library) is by far the fastest engine, so it is advised to keep the default version if you don't have your own engine setup.
References - top
[1] K. Schomburg, H.-C. Ehrlich, K. Stierand, M.Rarey; From Structure Diagrams to Visual Chemical Patterns, J. Chem. Inf. Model., 2010, 50 (9), pp 1529-1535